Probleemstelling:
Protein network and mutations
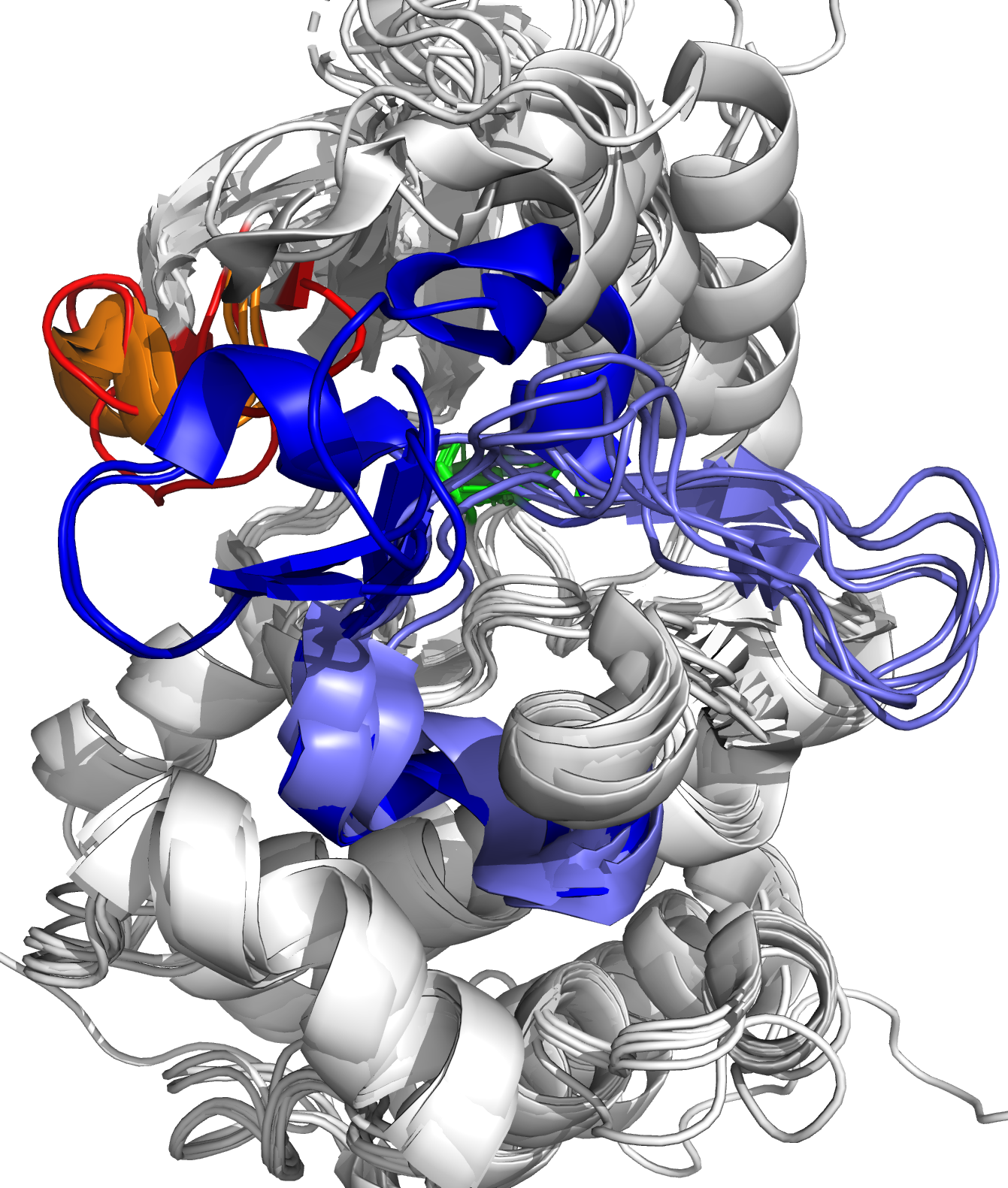
The human proteome consists of a tightly regulated network of proteins, where specific sites on these proteins allow them to interact with other proteins, small (drug) molecules, antigens, or ligands in general. The result of such binding interaction is a conformational change of the target protein, where it takes on a – sometimes slightly – different 3D structural position, which allows the target protein to project new binding sites, or to transfer functional groups from one protein to another. In other words, the binding interaction activates or inhibits a specific function of the target protein, which enables the interaction with yet another protein, and so on.
The structural information of proteins is encoded by their respective DNA sequences, where a mutation of a single DNA base pair can result in the formation of malfunctioning and/or misfolded proteins. Mutations are not necessarily harmful, as some DNA mutations result in the exact same protein product (DNA replicas), and some result in the substitution of an amino acid (AA) into a similarly sized AA with similar electrochemical properties. However, when a DNA mutation produces an AA substitution localized in/near an important binding site of the protein, it may no longer recognize its native ligands and disturb the protein signalling network, causing toxic and oncogenic cell-regulation.
Abelson tyrosine kinase, cancer and small drug molecules
The protein Abelson tyrosine kinase (ABL) is involved in cell differentiation, cell division, cell adhesion and stress response [1]â . ABL transfers a phosphate group from ATP to different substrate proteins, and forms an important link in a larger network of cell cycle regulators. Some patients develop the BCR-ABL fusion oncogene, where part of chromosome 22 is translocated to chromosome 9, resulting in a modified DNA sequence that codes for a fusion oncoprotein BCR-ABL [2]â . The oncoprotein BCR-ABL retains the functional ability of ABL, in fact, it performs too well. Indeed, while ABL is normally inactive (not transferring phosphate), the oncoprotein BCR-ABL is always active (transferring phosphate). This leads to increased cell growth and -division, and results in chronic myeloid leukaemia (CML, cancer).
Small drug molecules have been developed that artificially put BCR-ABL in the inactive state, by occupying the ATP-binding site of ABL. As such, ATP can no longer bind to ABL, and there is no phosphate group to be transferred. The first developed small drug molecule was imatinib, which proved to be very successful in combating CML, and is regarded as the first case of targeted drug design. Unfortunately, some patients have mutations near the ATP-binding site of ABL, which block the binding of imatinib. To this end, second generation drug molecules were developed. These were blocked again by mutations, spurring the development of third generation drug molecules.
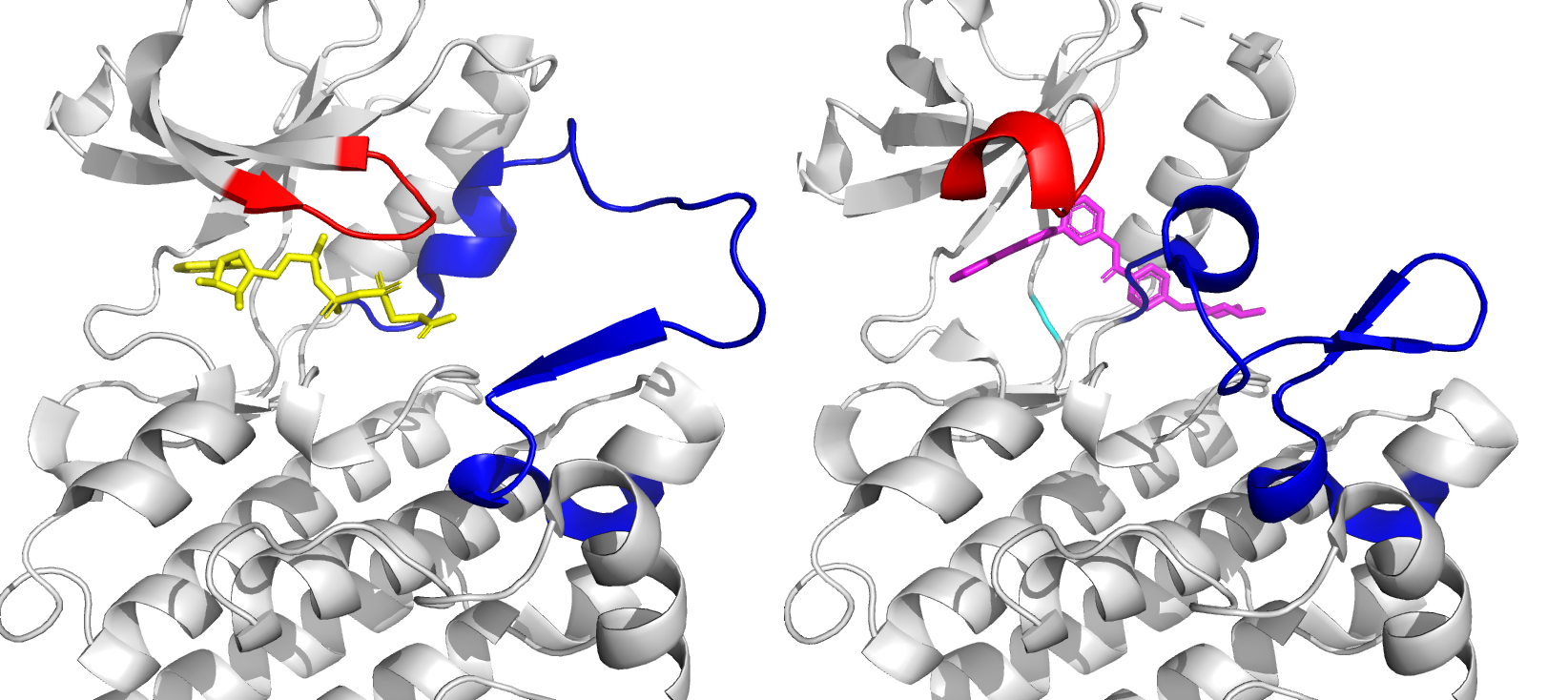
Figure 1: Close-up of ATP binding site of ABL kinase domain, with bound ATP (left, yellow) and bound imatinib (right, magenta). Imatinib inactivates ABL by competitive occupation of the ATP binding site. Important flexible loops are colored red (glycine-rich loop) and blue (activation loop).
Molecular dynamics for targeted drug design
While experimental techniques provide excellent methods to study proteins, they are time-consuming and expensive. Moreover, probing the proteins’ extremely fast configurational changes and interactions at the atomic scale remains challenging. To this end, computational techniques have been developed for high-throughput applications (e.g. protein-ligand docking in targeted drug design), and molecular dynamics (MD) simulations that provide the fully atomistic picture of proteins.