Probleemstelling:
Protein network and mutations
The human proteome is a complex network of proteins with specific binding sites that allow for interactions with other molecules such as proteins, drugs, antigens, or ligands in general. These interactions can cause a conformational change in the protein, altering its 3D structure and creating new binding sites. This can activate or inhibit the protein's function, leading to further interactions with other proteins. The structure of proteins is determined by their DNA sequences, and even a single mutation in the DNA can result in the formation of malfunctioning or misfolded proteins. While some mutations have no effect on the protein product or result in a substitution of an amino acid with similar properties, mutations that occur in or near important binding sites can disrupt the protein signaling network and lead to toxic and oncogenic cell regulation.
Abelson tyrosine kinase, cancer and small drug molecules
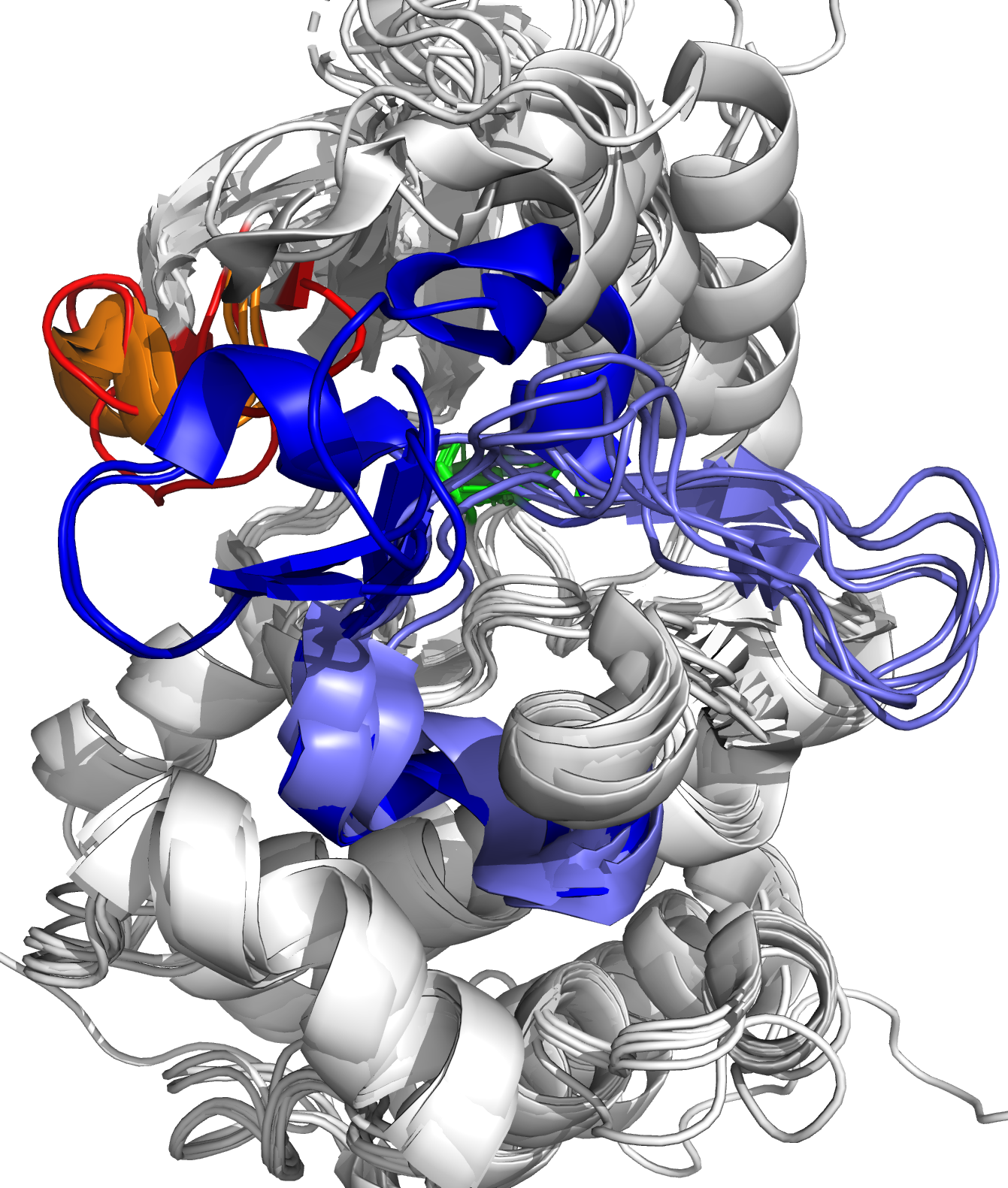
The Abelson tyrosine kinase (ABL) protein plays a critical role in cell differentiation, division, adhesion, and stress response [1]. ABL transfers a phosphate group from ATP to various substrate proteins and is an essential component of the larger network of cell cycle regulators. However, in some patients, a BCR-ABL fusion oncogene may develop due to a translocation of chromosome 22 to chromosome 9 [2]. This modified DNA sequence results in the formation of a fusion oncoprotein that retains the functional ability of ABL but is constitutively active, leading to increased cell growth and division and ultimately chronic myeloid leukemia (CML). The ATP binding site of ABL is shown in Figure 1. Small drug molecules have been developed to target BCR-ABL by artificially inhibiting its activity. These molecules occupy the ATP-binding site of ABL, preventing ATP binding and subsequent phosphate transfer. The first small drug molecule, imatinib, was successful in treating CML and marked a breakthrough in targeted drug design. However, some patients have mutations near the ATP-binding site of ABL, which prevents imatinib binding. This led to the development of second-generation drug molecules, which were also blocked by mutations, resulting in the creation of third-generation drug molecules.
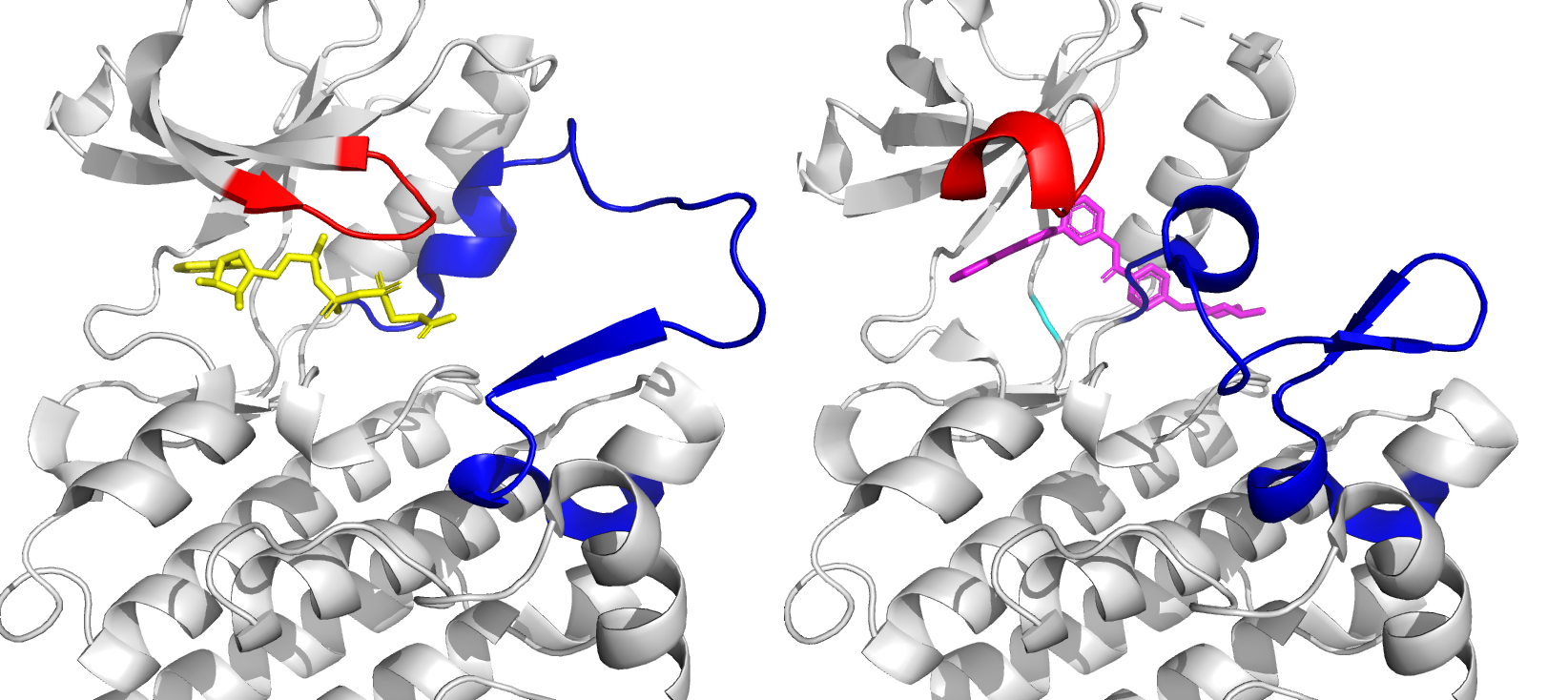
Figure 1: Close-up of ATP binding site of ABL kinase domain, with bound ATP (left, yellow) and bound imatinib (right, magenta). Imatinib inactivates ABL by competitive occupation of the ATP binding site. Important flexible loops are colored red (glycine-rich loop) and blue (activation loop).
New drug molecules that keep ABL in check
Drug molecules, such as imatinib, can occupy the binding sit on the ABL protein, and can keep it in an inactive conformation. To develop the next generation of drug molecules, it is a requirement that the drug molecule also retains ABL in its inactive conformation. The conformation of ABL is essential in the functioning of the drug. Therefore, an algorithm needs to be developed to detect what is ABL's conformation when it interacts with a new drug molecule candidate. In this thesis, ABL's conformation will be detected with simulations at the molecular scale.
Molecular dynamics for targeted drug design
Experimental techniques are valuable for studying proteins, but they can be costly and time-consuming. Additionally, the ability to observe and analyze the extremely fast configurational changes and interactions of proteins at the atomic scale remains a challenge. In response, computational techniques have been developed for high-throughput applications, including protein-ligand docking in targeted drug design. Molecular dynamics (MD) simulations are among these computational techniques and provide a fully atomistic view of proteins. MD simulations allow researchers to model the dynamic behavior of a protein at the atomic level over time. This provides insights into the protein's conformational changes and interactions with other molecules, such as drug candidates. By using MD simulations, researchers can better understand the binding mechanisms between proteins and drug candidates and predict how mutations may affect the binding affinity. This information can be used to design more effective and targeted drug molecules.